








General information
Home » Pharmacovigilance » General information
Pharmacovigilance is a set of activities related to collection, identification, evaluation, understanding, prevention and response to adverse reactions to medicines, as well as other problems relating to their use. Adverse reaction to a medicine is harmful and unintentionally caused reaction to a medicine, while an adverse event is unwanted experience created in the period of medicine administration for which the causality with medicine use does not have to be proven and represents any unwanted and unintended sign (e.g. an abnormal laboratory finding), symptom or disease, timely associated with medicine administration.
Unexpected adverse reaction of medicine is any harmful adverse reaction to the medicine that is not listed in the approved summary of product characteristics. Serious adverse reaction to a medicine is any adverse reaction that can cause death, is life-threatening, hospitalization or prolongation of hospitalization if for that there was no need before the usage of medicine, persistent or significant impairment or disability, congenital anomalies or distortions during nursing and other medically significant condition.
Pharmacovigilance system is a system established by marketing authorisation holder and the Institute, with the aim of monitoring the safety of medicines on the market and detecting any change in the benefit-risk ratio of their use. The benefit-risk ratio is an assessment of positive therapeutic effects of a medicine in view of the risk associated with its use.
Development and continuous improvement of the pharmacovigilance system is necessary since not all adverse reactions that can manifest during premarketing phase of development of the may be foreseen in the period before placing the medicine in question on the market. Reasons to it are as follows:
- Studies in animals cannot fully foresee safety of the medicine administered in humans.
- Subjects who participate in clinical trials are selected, conditions for the administration of the medicine in clinical trials are different from those in common clinical practice, the duration of clinical trials is limited and a relatively small number of subjects is included.
- Given that examined medicine is usually administered in less than 5000 patients prior to placing on the market, only adverse reactions that commonly occur may be noticed.
- At least 30,000 patients should take the medicine to make sure that we have not failed to notice adverse reactions whose incidence is 1 in 10000 (very rare)
- Information about rare adverse reactions, toxic effects of long-term use of the medicine, use in special patient groups (such as children, the elderly and pregnant women) or medicines interactions are either incomplete or unavailable.
Post-marketing safety monitoring of the medicine is particularly important for the detection of serious adverse reactions whose incidence is low and for adverse reactions that occur after long-term medicine administration, or after a certain period of time.
Owing to the information from the post-marketing period of the lifecycle of the medicine (spontaneous reporting, clinical trials, relevant literature, scientific journals, registers, etc.) we obtain true picture of safety profile of a certain medicine.
Based on collected information which are to be analyzed and assessed, the Institute, with the aim of protecting public health, may take certain measures prescribed by the Law on Medicines.
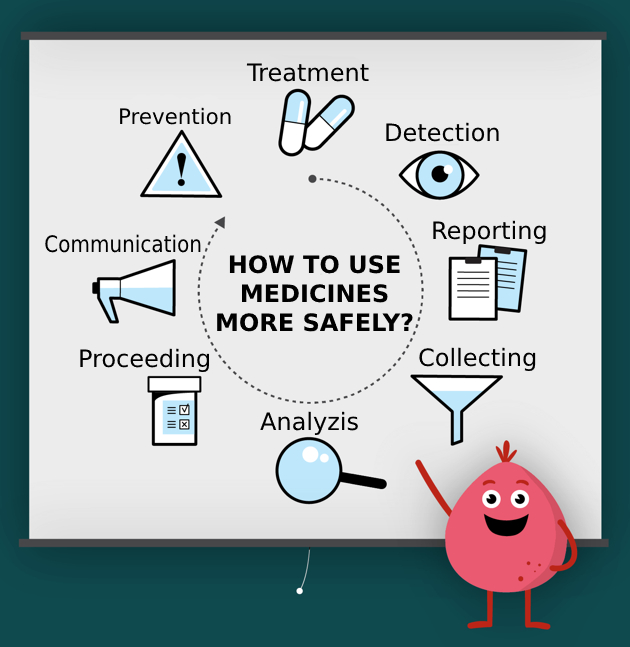
More information about pharmacovigilance and CInMED activities with the aim of promoting this scientific discipline is available in the section Publications and on our YouTube channel.
More information about the WHO Programme for international monitoring of safe use of medicines, which Montenegro has been a full member of since 2009, is available on the website of Uppsala Monitoring Centre in Sweden, independent organization that coordinates mentioned Programme: https://who-umc.org/.

Search register
Here you can search for medicines in our register

Sign up for
Newsletter
